Amplifying on the PCR Amplifier
PCR is an algorithm that amplifies DNA

Kary Mullis is not a theorist, but is a chemist who won the 1993 Nobel Prize for inventing the PCR method—an amplifier for DNA. He is also an alumnus of Georgia Tech, where I am these days, yet we do not always claim him as such—according to my friends in biology. While Kary is famous, and justly so, he has a quirky personality that does not always make everyone thrilled to be connected with him.
Today I want to discuss the details of his PCR method, which I hope you will enjoy. It really is an algorithm. Really. I think this is, at least partially, why there was such a controversy swirling around whether or not Mullis did enough to win a Nobel Prize. He certainly is one of the few who have been honored with a Nobel Prize for the invention of an algorithm; for example, Tjalling Koopmans and Leonid Kantorovich won their economics Nobel Prizes for linear programming in 1975. But there are few others. Also, PCR raises a pure theory problem that I hope someone will be able to resolve. More on that shortly.
Mullis invented PCR while he was employed at Cetus, now a defunct company. He got a $10,000 invention award, while Cetus eventually sold the patent rights for 300 million dollars. This was back when this was real money, even today that is a pretty good amount for a single patent. Mullis was not happy with the size of his award, which is not hard to understand.
This reminds me of a story about IBM. They have an award for the best invention of the year, which comes along with a nice, but relatively modest, cash award. Years ago the inventor of a certain encoding trick for improving the density of disk storage units was awarded this prize. Then, he was awarded the same prize the following year: for the same invention. This is like being “Rookie of the Year” for two years in a row. Since the invention made IBM billions of dollars in additional sales of disk units, they needed someway to thank him. So he got the best invention of the year, for the same invention, twice. Perhaps Cetus could have done something like this too.
Back to PCR. There was a huge court battle over whether the Cetus patent for PCR was valid or not. Several famous biologists, including some Nobel Laureates, testified against Cetus and Mullis: arguing that the PCR method was already known, that Mullis did not invent anything, and therefore that the patent should be thrown out. Mullis won.
I have read parts of the trial transcript and other accounts, and I think I have a theory why there was such outcry an against Mullis. First, he is—as I stated already—a character. If you have any doubts read his book—“Dancing Naked in the Mind Field.” Second, there was a great deal of money and prestige at stake. The patent, if valid, was extremely valuable. Finally, there was—and this is my two cents—the algorithm issue. Mullis invented no new chemistry, he discovered no new biological process, he found nothing new in the usual sense. What he did that was worthy of a Nobel was “just” put together known facts in a new way.
To me this is like arguing that the famous RSA algorithm is not new: we all knew about modular arithmetic, about primes, about Fermat’s Little Theorem, and so on. So how could Ron Rivest, Adi Shamir, and Len Adleman be said to have invented something? They did. As theorists, we appreciate the huge breakthrough they made—but the biologists not being oriented toward algorithms might have had trouble with PCR.
Let’s now turn to the Polymerase Chain Reaction (PCR) algorithm and the open question about it that I wish to raise. If you want you can jump to the next to the last section where I discuss the open question, but I worked hard on the next sections—the DNA diagrams were tricky—so at least look at them.
DNA
The cool thing about PCR is you do not need to know a lot about the deep structure of DNA in order to understand the method. There are a couple of key facts that you do, however, need to know. A single strand of DNA can, for us, be viewed as a string of four letters from the alphabet 

is an example of a single strand of DNA. The bonds denoted by the 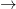
Two single pieces of DNA can form a double strand of DNA, which consists of two such sequences placed together: one running left to right, the other right to left, and where the letters meet they follow the two famous Watson-Crick rules:

The 

Thus, the following is an example of a double strand of DNA:
| A | |
T | |
G |
| | | | | | | ||
| T |  |
A | |
C |
Note, the top strand goes in the opposite direction of the bottom strand.
Mullis used a few simple operations that can be done to DNA. One is that DNA can be heated. When a double strand of DNA is heated properly (not too hot, and not too fast) it breaks nicely into the top and bottom strands. Thus, the above double strand would break into:

A key point is that

are the same.
Another simple operation is that DNA can be cooled. In this case single strands will try to find their Watson-Crick mates and bond. Note, the above two strings could reform again into,
| A | |
T | |
G |
| | | | | | | ||
| T | |
A | |
C |
The key is could. In order for this to happen they must find each other. If they were alone in a test tube, it would be very unlikely to happen. But, if there are many copies of each type, then many of the above double strand would form.
The last operation that Mullis uses is the most interesting, and it relies on the enzyme polymerase. Consider the following piece of DNA made of two single strands,
| A | |
T | |
G | |
G | |
G |
| | | | | |||||||
| C | |
C | ||||||
Note, that the bottom is incomplete. How do we get the remaining part of the bottom strand constructed? The answer is to add the enzyme polymerase and a sufficient supply of each nucleic acid 
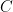

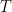
| A | |
T | |
G | |
G | |
G |
| | | | | | | | | | | ||||
| T | |
A | |
C | |
C | |
C |
An important point: polymerase only completes the bottom if it is already started. Thus, a single strand of DNA would not be changed at all. The top must have some of the bottom started already: actually the bottom part must be at least a few bases. One more key point to make: polymerase only adds in the direction of the DNA strand. Thus, in this case nothing will happen:
| A | |
T | |
G | |
G | |
G |
| | | | | |||||||
| T | |
A | ||||||
All these steps—heating, cooling, and polymerase—were known for decades before Mullis’s work; amazingly that is all he needs to do the PCR algorithm.
Basic PCR
I would like to explain what I will call the basic method first, then I will explain the full method. Suppose that you start with a double strand of DNA of the form:
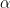 |
|
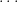 |
|
 |
| | | |
| | ||
 |
|
|
|
 |
where 
Then, all you do is heat, cool, heat, cool, for some number of cycles. The magic is that this is enough to make an exponential number of copies of the original DNA. To see this consider what will happen after the first heat and cool cycle. The above will become:
| |
|
|
|
|
| | | ||||
| |
||||
and
| |
||||
| | | ||||
| |
|
|
|
|
This follows since during heating the double strand breaks into the top and bottom strands; then during cooling the most likely event is for the abundant primers to attach to the top and the bottom. It is very unlikely for the double strands to re-attach together—since there are so many primers. Next, polymerase does its magic and after a while the above becomes:
| |
|
|
|
|
| | | |
| | ||
 |
 |
 |
|
 |
and
| |
|
|
|
 |
| | | |
| | ||
| |
|
|
|
|
Thus, we have doubled the number of strands of the DNA—we have an amplifier.
Full PCR
The full PCR method can copy a selected part of a long strand of DNA. This is really the method that Mullis invented. The ability to copy only part of a much longer strand is of huge importance to biologists. Curiously, it is exactly the same method as the basic one. The only difference is the analysis of its correctness is a bit more subtle: I find this interesting, since this happens all the time in the analysis of our algorithms.
Suppose that you start with the following double strand of DNA:
 |
|
|
|
|
|
|
|
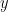 |
| | | | |  |
| | | | ||||
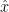 |
|
|
|
|
|
|
|
 |
As before, put the following all together in a test tube. The double strands of the above DNA; lots of single copies of
| |
|
|
|
|
| | | |
| | ||
| |
|
|
|
|
Thus, only the DNA between the primers is exponentially increased in number. Other, partial pieces of DNA will grow in quantity, but only at a polynomial rate. This is the PCR algorithm.
Can PCR Amplify One Strand of DNA?
Can PCR copy one piece of DNA? I have asked this over the years of my biology friends and have been told that they believe that it can. However, usually PCR is used to amplify many strands of DNA into a huge amount of DNA. So the problem that I want to raise today is: can theory help determine the ability of PCR to amplify even one piece of DNA? The obvious lab protocol would be to prepare test tubes that have exactly one piece and see how the PCR method works. The trouble with this is simple: how do we make a test tube that is guaranteed to have exactly one piece of the required DNA? I believe that this is not completely trivial—especially for short strands of DNA.
The Amplifier Sensitivity Problem
Note, even if biologists can prepare test tubes with one strand, the following problem is interesting since it could have applications to other amplification methods. The problem is an ideal version of trying to decide if PCR can amplify one strand or not, without the ability to create test tubes with one strand.
I have thought about this issue some, and would like to present it as a pure theory problem. I will give a simple version, but you can probably imagine how we could make the problem more realistic.
Imagine that you have boxes—think test tubes. A box can have a number of balls—think DNA strands—inside: the number can range from 
However, you do have a detector, that given a box does the following: if the box has less than 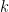
Your task is to determine the value of 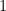
- You can ask for a new box. This will be filled with a varying number of balls, but the number of balls
in the box will always be vastly larger than
- You can ask that the contents of a box be randomly divided into
boxes. This operation destroys the original box and returns to you
and the division is random.
That is all you can do. The question is: can you devise a protocol that will get a good estimate on the secret value of 
Open Problems
The main question today is to solve the amplifier sensitivity problem and find 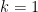

Note, even if PCR has been shown in the lab to be able to amplify one piece of DNA, these protocols may be of use in the future with other types of amplifying mechanisms. As science moves down into the depths of nano scale I can certainly imagine other applications of a protocol that can determine how sensitive an amplifier is.
Please solve these problems. Or prove that they cannot be solved.


What do you we know about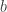 , the quantity of balls in a new box? Is it constant? Distributed according to a known/unknown distribution?
, the quantity of balls in a new box? Is it constant? Distributed according to a known/unknown distribution?
If I understand your problem statement, the answer is not well-defined since if you double both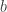 and
and 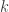 the system will behave approximately the same. Maybe I misunderstood the available primitives?
the system will behave approximately the same. Maybe I misunderstood the available primitives?
I think that is too simple an argument. There is a pigeonhole type principle lurking about, and 1 vs 2 will not behavior the same way.
I am not a biochemist, but it seems to me that a major feature of PCR not accounted for in the given model is that if you do try to use PCR on a sample with no DNA strands, I don’t think you’ll end up with DNA strands (although if that’s wrong, please correct me!)
In that case, it certainly does seem possible to devise an algorithm to distinguish between k = 1 and k = 2, working roughly as follows: repeatedly split the balls between two boxes and feed one to the detector, until the detector detects no balls (say after m repetitions). Then I believe we can get a reasonable bound f(m) on the expected number of balls in the remaining box; split this box between n >> f(m) boxes, and if k = 2, w.h.p. the detector will detect balls in none of them.
There are several obvious flaws in the above approach, but I think they may be able to be smoothed over. Still, this is a typically great post (and I was just reading about Kary Mullis yesterday! He certainly is…unusual. A particularly ironic example is his skepticism of the belief that HIV causes AIDS, when PCR techniques have been used to refute some of the claims of the HIV/AIDS denialists over the past decade.)
You are right. If no strands get none. I thought I said that. I think your approach is the right idea, but the details seem to be a bit tricky. I believe we can make something work, but feel free to try and see if can get exact bounds.
I decided not to add any details on his strange beliefs—I try to always be positive. However, you are dead on about the irony.
When you say say the balls are distributed in a uniformly random manner into the boxes, do you mean that each ball’s placement is uniformly distributed, with the placement of each ball being independent? Or do you mean some more sophisticated distribution like uniformly over all possible partitions of the balls into boxes? I’m not sure if this makes much of a difference or not, but I thought I ought to at least ask.
I meant this: If the box has b balls and there are three boxes, then each ball in thrown into one of the boxes with a random choice. Does that help? Also I am not sure will matter much.
Yeah, that makes sense. If you don’t terribly mind, I’ll be explicit about what I meant earlier.
Say you have a box with two balls and you ask for it to be split into two boxes. You could count that there are 4 ways of doing this: 12| 1|2 2|1 |12 Ball 1 can be placed in the first or second box, and similarly with ball 2. If we think of these as the 4 possible cases, then the probability of both balls being placed into the first box is 1/4. This seems to be what you meant.
Alternatively, it would seem reasonable in this sort of a case to consider the balls as indistinguishable from each other, in which case I might count there being 3 ways to place the two balls into two boxes: **| *|* |** If I specify a uniform distribution over these cases, then the probability of both balls being placed into the first box is 1/3. I suppose these wouldn’t quite be partitions (ordered partitions, perhaps?) of the balls, but this is certainly a different distribution.
I suspect it wouldn’t make too big of a difference. It’s just the usual first question that pops into my head when someone says “uniform random.” =)
the two distributions are the same (think symmetry).
Harrison: “repeatedly split the balls between two boxes and feed one to the detector, until the detector detects no balls”
For a detector of sensitivity k, let p(n,k) be the probability that, starting with a box containing n balls, at the end of Harrison’s process the number of balls in the box not fed to the detector is = k, p'(n, k, m) also converges on some non-zero value as n goes to infinity. If so, then there should be some way of deriving some quantities we can measure which will distinguish different values of k. (E.g. the probability that if you divide up the remaining box into a boxes, the detector will detect balls in b of these boxes.)
Thank you for the post. DNA diagrams were very helpful. I have read about about PCR after watching ‘s TED Talk (http://www.ted.com/talks/kary_mullis_next_gen_cure_for_killer_infections.html), but diagrams cleared my mind.
It isn’t hard. For sufficiently large values of the number of boxes (L), the fraction of “yes” answers will be proportional to L^-k.
In 1905, Albert Einstein solved a somewhat similar problem. In Brownian motion you have many very small events (air molecules colliding with the test particles), and you can observe their sum over relatively long intervals. The problem was to determine the total number of collisions, from which it was possible to to calculate (among other things) the value of Avogadro’s number. Counting the actual *number* of atoms was important at the time, because the very existence of atoms was still controversial.
A far more difficult problem is to find a protocol that finds k while consuming the smallest possible sample.
A story similar to that of César Milstein and Monoclonal Antibodies:
The journey of monoclonal antibodies all started when an Argentinian émigré called César Milstein arrived at the Laboratory of Molecular Biology in Cambridge, the same laboratory where Francis Crick and James Watson discovered the structure of DNA in 1953. It was to be here that Milstein, together with Georges Köhler, pioneered the seminal technique for the production of monoclonal antibodies in 1975 and showed their clinical application for the first time…